以下文章来源于CC周刊 ,作者张洪涛
撰文 | 张洪涛(宾夕法尼亚大学医学院副教授)
● ● ●
- 核心提要 -
1. 虽然中国对于 “冠状病毒” 的注册临床试验有589个(截至2020年5月20日),但其中有效的2期与3期干预性临床试验共只有8个。除了疫苗之外,证明药物疗效的临床试验,近乎全部 “翻车”。
2. “临床试验” 不能仅靠行政审批,要依靠科学临床试验的“循证审批”。注重质量而非数量,才能成为真正的制药强国。
3. 五大原因降低临床试验质量:回顾性研究过多;单臂实验阻碍无对照;临床试验主要终点选择草率;国内试验样本量较少;而临床失败的最大原因是没有特效药,只能反复试验,选择联合疗法。
4. 中国要做得更好,需要充分利用体制优势,推行多中心重大试验,扩大实验样本数量;取舍无前景的临床试验;组织临床试验的人专业化。
01 589项新冠试验 是不是都“失败”了?
中国开展了589项新冠临床试验,但疫情在3月底已基本结束,到目前为止,除了疫苗的临床试验,能做的已经做完,但似乎没有拿得出手的结果,是不是都 “失败” 了?是中国的临床试验出了什么问题吗?
在中国正式的临床试验,都会在 “中国临床试验注册中心” 登记注册。5月20日,在注册中心的网站上,使用“冠状病毒”进行检索,显示出有589个已经登记的临床试验。
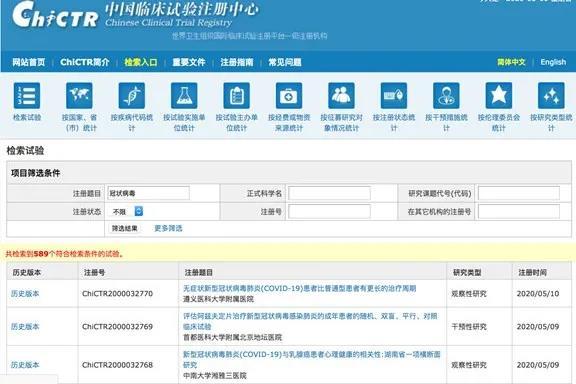
在解读临床试验之前,有必要正确地区分一下不同临床试验的目的。并不是所有的临床试验,都是为了探索新的治疗方案。有一些临床试验,只是对患者进行观察,或者获取患者的一些病理样本,进行分析研究。涉及治疗的,属于干预性研究。在这些注册的新冠临床试验中,明确注明为干预性临床试验的,有310个,大约占所有相关临床试验的一半。
而在这些干预性的临床试验中,大部分为早期临床试验,属于预试验,主要目的为探索可行性及治疗的安全性,目的本来就不是为了获得疗效的证据。也有一些是疫苗的临床试验,参加试验的并不是新冠患者,不在本文的讨论范围。
大家所关注的,是那些能够证明药物治疗效果的临床试验,也就是临床2期、3期的试验。这样的试验有多少呢?
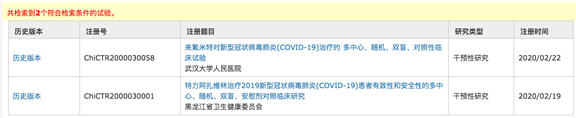
在注册中心正式登记的临床试验中,3期临床试验只有2个:
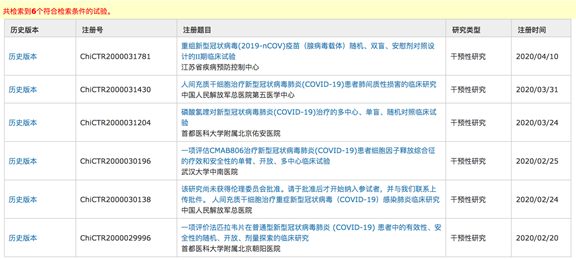
2期临床试验,也只有6个:
2期和3期的临床试验,总共加起来只有8个。(注:曹斌教授所负责的瑞德西韦、克力芝两项临床试验,直接在clinicaltrials.gov注册,不在这289个临床试验之中)
除此之外,还有56个被列为 “上市后研究/4期临床” 的试验,也应该是为了证明疗效的。这也比较奇怪,理论上来说,这一类研究所指的 “上市”,是针对该适应症而言,但是很显然,即便某个药物已经获批其他的适应症,但在申报新冠临床试验的时候,该药物也不会有 “新冠” 这个适应症,就不存在已 “上市” 的问题,也就不可能有所谓的4期临床。
以癌症治疗为例,假设一个获得药物批准用于黑色素瘤,如果想进行一个肺癌的临床研究,最多也就只能是进行3期试验,不可能进行4期临床。同样的道理,虽然氯喹被正式用于疟疾的治疗,但是并不能认为就可以用来进行治疗新冠,所要进行的临床试验,也不可能是4期临床试验。
不管怎样,从注册登记就可以看出临床试验的混乱。也可以看出在这些临床试验里,有着太多太多的凌乱的预试验。一场人类经历的瘟疫,变成了临床试验的 “盛宴”,结果感觉只留下了杯盘狼藉。
到目前为止,中国的疫情已经基本结束,中国的临床试验也已经基本结题,但似乎并没有出现什么值得欢呼的好消息,这些证明药物疗效的临床试验,可以判定近乎全都 “翻车” 了。
早在2月24日,《中华流行病学杂志》就发表了《关于科学、规范、有序地开展新型冠状病毒肺炎相关临床试验的建议》。专业人士指出,因项目众多,患者数量不能满足研究要求,这会导致研究难以获得预期结论,临床研究也难以提供高质量的有效性和安全性证据,会让受试患者、研究者和管理部门的努力付诸东流。
而在3月29日,国务院应对新型冠状病毒肺炎疫情联防联控机制科研攻关组也发布了一份 “关于规范医疗机构开展新型冠状病毒肺炎药物治疗临床研究的通知”,要求对有关的临床试验加强监管,并要求 “自发布之日起开始实施。已经开展(首例受试者已入组)但尚未完成的临床研究,医疗机构应当自本文发布之日起3个工作日完成立项、登记并上传信息等工作。逾期未完成的医疗机构,不得继续开展临床研究工作。”
根据报道,在此之后,有43项临床试验已主动撤回。但是,中国的疫情在2月底基本已经进入收尾状态,能做的临床已经做了,不能做的也基本没有机会做了。
疫情就是一面镜子。这次疫情,暴露了国内临床试验的“大跃进”以及一轰而上的混乱局面,所带来的严重后果:虽然有589项临床试验,但除了疫苗之外,至今尚无一项拿得出手的成果。

02 临床试验的目的到底是为什么?中国药品质量为何不如印度?
临床试验,是为了让一款药物获得批准,能够正式推广和销售吗?
如果这是真正的目的,那其实有很多办法可以达到这个目的,甚至并不一定要通过非常复杂的临床试验。比如说,“非典” 发生于2003年,在那一年及随后的几年里,中国快速批准了大量的药物。有资料表明,仅2005年一年,药监局批准了11086件药品注册申请事项,其中批准新药1113个,改变剂型的品种1198个,仿制药品8075个。
这个数字是惊人的,且不说仿制药品,就算新药,也几乎是美国每年批准的新药数量的10倍。
当年获得批准的那些 “新药” 都还在,但是,当年的药监局局长郑筱萸,已经因为受贿被判处死刑,并于2007年被执行注射死刑。
当年的那些 “新药” 到底有没有做过什么临床试验?结果有多可信?我想答案已经被郑筱萸带走了。
所以,做临床试验的目的,并不是为了让一个药获得批准,而是要知道一个药物是否治疗有效、安全可靠。这是一个科学问题,就必须要遵循科学的方法来回答。
至于药品的审批,这是一个行政问题。从理论上来说,如果审批基于科学的临床试验,可以减少 “郑筱萸” 的出现。但如果纯粹是靠行政手段来审批新药,“郑筱萸” 就不可避免。
如果说2007年之前,是中国药监审批的 “至暗时代”,那在2007年之后,中国一直在“循证审批” 的道路上前进。为了解决历史遗留的包袱,中国推行了药物的“一致性评价”,要求仿制药品要与原研药品质量和疗效一致。如果仅靠行政审批的手段,中国的药品可以在数量上迅速大跃进,“赶英超美”,但是在质量上连印度都无法超越。如果坚持“循证审批”的道路,虽然速度会慢一点,但是可以薄积厚发,真正成为一个制药强国。
应该说,“临床试验” 的观念在中国还是比较深入人心的,也正是因为如此,在新冠爆发之后,才一下子冒出几百个临床试验。从一个方面来说,这是对“循证医学”的鼓励和期望,是值得点赞的;但是从另外一个方面,却暴露出了各方人士对临床试验的认知不足。
武汉金银潭医院的医务人员与患者。
03 为什么将近300多项治疗性临床试验,不可能得到治疗的有效性数据?
看来临床试验里有很多坑。进行临床试验,需要注意哪些问题?或者说怎样做,才能提高临床试验的质量,提高获得成功的胜算呢?
1. 回顾性研究,并不是真正的临床试验
在很多人的印象里,临床试验就是人体试验,只要是有人体试验中的数据,就当成是临床试验的数据。这是错误的。
这里涉及一个 “前瞻性” 和 “回顾性” 的问题。“前瞻性”,是指在研究开始的时候,还没有开展干预性治疗;而 “回顾性”,是指在研究开始的时候,干预性治疗已经结束,因为只是对之前结果的分析。
比如登记注册的一个干预性试验 ,项目名称为 “大剂量静脉注射维生素C在新型冠状病毒肺炎(COVID-19)重症患者急性加重期的改善作用研究”(注册号:ChiCTR2000032716)。这个研究里,分别有6个重症患者,6个危重症患者。但是,这个研究虽然是干预性研究,但是却是 “回顾性研究”、“病例研究”,即便发现其中部分患者好转了,也不可能得出治疗有效的结果。
为什么呢?因为 “回顾性” 的研究,可能只是选择某一个有局限的人群,看到的只是森林里的几棵树,可能有代表性,也可能没有代表性。如果开展 “前瞻性” 的临床试验,才能比较客观地获得结论。
之前媒体上曾经提到过一个《中华传染病杂志》发表的克力芝和阿比多尔的临床使用的报告,称其为临床试验 [1]。该研究未发现洛匹那韦利托那韦和阿比多尔具有改善新冠肺炎症状或缩短呼吸道标本病毒核酸转阴时间的作用。但是,这份报告只是一个回顾性分析,并不是严格设计的临床试验。这样的研究,只能推断出药物的治疗效果可能不明显,并不能说明是否就毫无效果。
与此相反,媒体上还有一个有关中药治疗新冠肺炎的报道:
“武汉市中西医结合医院住院1476例,其中重症、危重症患者662例(这当中中药汤剂组484例,非中药汤剂组178例)。中药汤剂组死亡15例,未用中药汤剂组死亡56例。他介绍,中药汤剂组的死亡风险下降了87.7%,与未用中药汤剂组的差异具有统计学意义。核算死亡率可知,中医汤剂组死亡率3.1%(15/484),非中药汤剂组死亡率31%(56/178),两者相差高达10倍。”
这也同样属于回顾性分析,并不是临床试验。如果是临床试验,在分组进行治疗的时候,会保证 “中药汤剂组” 和 “未用中药汤剂组” 两组患者的病情、年龄都有可比性。否则,有可能危重症患者已经做上了插管,连自主呼吸都无法进行,当然也无法喝下中药汤剂,所谓的“未用中药汤剂组”,其实是因为病情更加严重而死亡率比较高,跟是否喝了中药没有多少关系。
如果研究者认为回顾性研究的结论可靠,需要进行正式的 “前瞻性” 临床研究来证明。
2. 没有对照组的临床试验 (单臂试验),无法得出结论
既然是为了获得疗效的证据,临床试验就需要有对照治疗。有了对照治疗作为参考,才知道新的治疗到底有没有更好的效果?如果对照治疗是安慰剂,新的治疗看不到更好的效果,就说明用不用新的治疗,区别不大。
现代医学要前进,临床试验不但需要有对照,而且必须使用目前已经证明有效的标准治疗,以保证新药创新是真的创新,不是原地踏步。
只有对于一个有100%死亡率的疾病,如果一个药物可以减少死亡率,从伦理上考虑,可以不使用对照治疗。但是,新冠肺炎并不是100%致死的疾病,甚至有无症状的人,如果没有对照治疗,确实不知道某种治疗的效果到底如何。
在临床登记中,有23个干预性试验登记为单臂试验。有一个研究, “评价达诺瑞韦钠片联合利托那韦治疗新型冠状病毒肺炎(COVID-19)患者的疗效及安全性的随机、开放、对照临床研究 ”(注册号:ChiCTR2000031734)。即便课题的标题里有 “对照试验”,但是试验设计里只有一组:
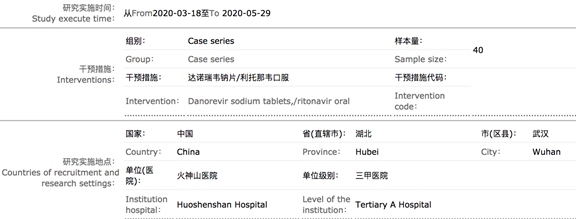
需要说明的是,对于一个未知的药物,确实有必要进行临床一期研究,以获得药物使用剂量和安全性方面的信息,这种早期的研究是不需要对照的,但是其目的并不是为了验证是否有药效。
3. 为何有的临床试验不用死亡率而用 “减少病毒转阴时间” 做为终点?
临床试验的主要终点,需要谨慎选择
在设计临床试验的时候,必须确定一个考察的主要指标,如果治疗方案在这个指标上显示出明显优势,就可以说临床试验“达到了主要终点”,获得了成功。由此可见,对这个指标的选择,是很关键的。
对于新冠病毒感染来说,这个指标可以是病毒的转阴时间,也可以是临床症状的改善,还可以是死亡率。
很显然,如果入组临床试验的轻症患者比较多,死亡率就不是一个好的指标,因为即便对照组死亡率也不会太高。理论上来说,病毒转阴的时间,就是一个比较合理的指标,很多临床试验也是使用“减少病毒转阴时间”作为临床试验的主要终点。
但是,由于病毒核酸检查存在假阴性的问题,不管是由于取样操作还是试剂盒的原因,报道中假阴性的比例可以高达40%。对于一个50人的临床试验来说,有20人可以在一次检测中出现假阴性,有8人可以连续两次检测出现假阴性,成为误诊。如果以两次转阴作为主要指标,很显然这8个误诊将极大地干扰试验的结论。
国内和国外所开展的瑞德西韦临床试验得到了不同的结论,虽然两个试验的主要终点是临床症状的改善,但是改善的具体标准是不一样的。在国际试验中,最后使用也并不是一开始所制定标准,而是后来修改过的。有人对此可能会惊讶,感觉研究者有投机取巧的嫌疑。实际上,这是允许的,只要是在揭盲[Patricia2] 之前进行修改,理论上都不影响研究的结论。当然了,在一般情况下,都需要与FDA提前沟通,在获得认可后修改主要终点,否则研究数据也会受到质疑。
4. 临床试验的样本量,决定了瑞德希韦等试验导致的国内外不同结果
如果要比较山的高度和人的高度,因为差距太大,只需要比较一座山、一个人就行了;如果要比较两个国家的居民的身高,就不能随便找几个人来比较,以此获得靠谱的答案。
临床试验到底需要多少人?这取决于试验组和对照组的区别有多大。
国内洛匹那韦/利托那韦对重症新冠肺炎的临床试验,得到了阴性的结果[2]。洛匹那韦-利托那韦组和常规治疗组在入组治疗28天时的死亡率分别是19.2%和25.0%,从数据上看,两组还是有一点区别的,洛匹那韦-利托那韦组的死亡率要低一点,但这差别在统计分析上达不到显著性。
如果是19.2% 和25.0%的差别,理论上需要多少受试者才能看出洛匹那韦-利托那韦的治疗效果呢?1600人。这个临床试验实际上入组了多少人呢?199人。这1600人和199人的差距,就是失败和成功的差距。
我们再看瑞德西韦临床试验。国外的试验获得了成功,总共入组了1063人。中国进行的试验夭折了,自然没有获得一个阳性的结果,入组了多少人呢?236人,只到达了预期入组人数的一半。
在中国的310个干预性新冠临床试验中,并不是所有的项目都有对照组。在那些有对照组的临床试验中,有多少入组人数是基于对统计学方面的考虑?我相信这会是一个悲观的答案。
前述曾提及一个注册登记的临床研究, “高剂量维生素C联合中药方剂治疗普通和重型新型冠状病毒肺炎(COVID-19)的疗效及安全性”(注册号:ChiCTR2000032717),根据所提交的计划,该研究将总共入组60名患者,但是要分6个组,每组只有10人:
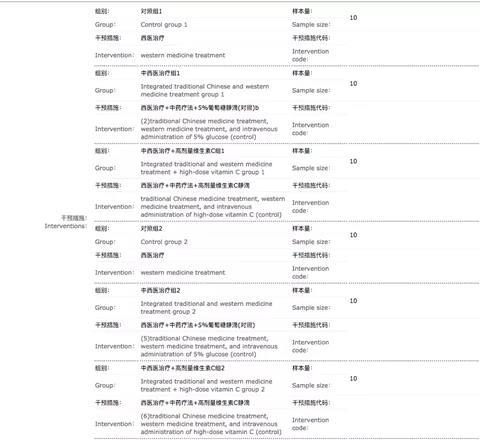
像这样的研究,比比皆是。
在疫情中,世界卫生组织联合考察组在中国进行了考察,在对中国防疫工作努力做出肯定的评价之后,也在如火如荼的“临床试验”中发现了问题:本来新冠病毒的感染者很多,但是随着大量患者出院,而同时展开的临床试验太多,招募患者变得越来越难。
在中国进行考察时,WHO联合考察组外方组长、世界卫生组织总干事高级顾问布鲁斯·艾尔沃德认为唯一有希望的是瑞德西韦的临床试验。当时所发表的观点,被很多人误读了,觉得WHO的官员也很不靠谱,试验结果还没有出来,怎么就敢给药物的有效性背书?实际上,他这个说法想要表达的,并不是对瑞德西韦有效性的支持,而是对绝大部分在开展的临床试验的失望,从设计上就可以看出失败的苗子,因为根本不可能获得是否有疗的确切答案。
5. 临床试验失败的最大的原因:试验药物疗效不明显,没有特效药
在导致临床试验失败的所有原因中,药物的疗效不够好,应该是最重要的原因。
如果瑞德西韦是一个特效药,治疗效果与对照组的差距,是山高与人高的差距,就不会在乎到底是入组了400人还是200人,甚至像很多试验设计的那样,40~50人就足够了。
如果瑞德西韦是一个特效药,也不用在乎临床症状改善的标准,是用中国研究中使用的标准,还是国外研究中的标准,更不需要小心翼翼地对标准进行修改,尽量把药物治疗的优势展示出来。
很不幸的是,我们面临的就是一个没有特效药的时代。洛匹那韦/利托那韦是抗艾滋病毒的药物,并不是为新冠病毒量体裁衣而设计的药物;瑞德西韦是抗埃博拉病毒的药物,也不是为新冠病毒量体裁衣而设计的;氯喹是抗疟疾的药物,同样也不是为新冠病毒量体裁衣而设计的。
正是因为在临床研究过程中发现药效不行,才有必要对试验进行调整。当国外瑞德西韦临床试验扩大入组人数规模时,其实就可以了解到瑞德西韦并没有特效药的效果。如今美国FDA给予瑞德西韦紧急使用授权,批准用于新冠的治疗,也不认为它是一个特效药。但是,瑞德西韦可以成为 “鸡尾酒” 治疗[Patricia3] 中的一个部分,联合其他药物来获得更好的效果。
“三个臭皮匠,顶个诸葛亮”。所以,在没有特效药的时代,联合疗法就是一个必然的出路。国内的洛匹那韦/利托那韦临床试验失败了,但是,一项来自香港大学的2期临床试验结果显示:使用 “干扰素β1b+利巴韦林+洛匹那韦/利托那韦(克力芝)” 的三联方案治疗,可以加快轻中度新冠患者的康复!这项研究发表在5月8日的《柳叶刀》上 [3]。
这个研究的主要考察指标,是鼻咽拭子的病毒转阴时间,从开始治疗到连续两次核酸检测阴性,使用三联方案治疗只需要7天,而使用洛匹那韦/利托那韦的对照治疗需要12天。当然,从治疗效果来说,三联方案越早使用,效果越好,在症状出来后7天之内开始治疗,效果非常明显,但是如果在7天以后,因为担心干扰素β1b可能导致免疫过激反应,治疗方案只是利巴韦林+洛匹那韦/利托那韦,治疗效果就不明显了。
04 对中国的临床试验研究的三点建议
临床试验的效果不佳,很容易进行全盘否定。但是,临床试验是科学实验,严格来说,只要是认认真真的试验,没有绝对失败的试验,只有尚未成功的试验。即便治疗组的疗效比对照还差,也可以证明这是一条错误的道路,需要走其他的治疗方式。
虽然科学研究都是有意义的,但是面对着对人类影响巨大的传染病,当务之急是尽快找到有效的治疗方案。
病毒给人类带来了灾难,其实也带来了机会,但如果不能珍惜机会,那才是人类最大灾难。怎样抓住机会,仁者见仁,智者见智。
复盘中国进行的300多项 “治疗性” 新冠的临床试验(这300多项为明确标注干预性治疗新冠病毒的临床试验,其他的200多项为回顾性等形式的临床试验),也许更有意义的问题应该是:我们如何可以做得更好?
做为一个在美国从事临床研究数十年的 “局内人”,我认为如下几点,应该可以作为抛砖引玉的建议:
1. 需要充分利用中国体制的优势,集中力量推行有前景的多中心重大试验
前面讲过,对于临床试验来说,样本量是决定试验是否能获得阳性结果的一个关键因素。新冠爆发后,武汉就有几万确诊患者,大家感觉入组人数不是很多,但是中国瑞德西韦招不满患者的事情确实发生了。
并不是所有的患者,都适合一个药物,而所有临床试验,都会设置患者入组的条件。在某一个医院能入组试验的患者,其实非常有限。如果仅靠研究者发起临床试验,局限于研究者所在的医院,无法获得足够的样本量。
也正是因为如此,目前国际上新药的临床试验一般都在多中心开展,背后都需要有制药企业的支持。多中心开展临床试验,如果成功了,也更能说明治疗方法的普适性。
反观目前新冠的治疗药物,大部分都是老药新用,很多都是没有专利保护的药物,很难由药企来支持临床试验的费用。这正是因为这个原因,300多个新冠的临床治疗试验,几乎都是研究者发起的试验。
中国在抗疫过程中展示出来的社会组织能力,已经为世人所瞩目。如何充分利用政府、社会对研究的支持,进行有效的协调,组织大规模的临床试验,而不是规模小、得不到明确答案的试验,国内相关主管部门应该有这个智慧。
值得一提的是,香港获得阳性结果的洛匹那韦/利托那韦三联治疗临床试验,在香港6所公立医院开展,总共入组了127名新冠肺炎患者。这听上去没有多少,但是人数已经是当时香港全部确诊患者的80%。试想一下,如果不是集中力量,选择重点进行的临床,这个试验还可能成功获得阳性结果吗?
2. 如何取舍那些没有前景的临床试验:抗病毒成分半抑制浓度是100 uM以上,基本可以不批准
如果要集中力量,那就不得不有取有舍。取舍的标准是什么呢?应该也是以实验数据为准则。
在体外实验中,瑞德西韦、氯喹的体外抗病毒活性(半抑制浓度)分别是0.77 uM 和 1.13 uM[Patricia4] 。这个数值越低越好,说明药物只要很低的浓度,就能达到抑制效果。
一般来说,只有体外试验的数据,根本不能用来作为支持临床试验的证据。即便是一个药物已经用来进行其他的临床治疗,也只能提供安全性方面的证据,并不能保证临床试验就可以获得成功。这就像一个获得批准用于黑色素瘤的抗癌药物,如果没有进行肺癌的临床试验,也不知道是否就可以用来有效地治疗肺癌。
但是在面对疫情的特殊情况下,根本没有时间先进行动物实验来验证,只能依靠体外细胞试验的数据来作为是否进行临床试验、是否优先选择的依据。
目前临床试验的结果证实,即便是uM级别的活性物质,临床抗病毒效果也非常有限。所以,如果一个 “神奇” 的抗病毒成分,半抑制浓度是100uM以上,那还是洗洗睡吧,千万不要去占用临床资源,去抢夺病人。
有时候,不作为,是对社会最大的贡献。

3. 专业的事,让专业的人来做
不可否认,临床的医生有很多的治疗经验,这是必须尊重的。但是,这并不意味着任何一个临床医生,都可以专业地领导进行大规模的临床试验。组织这样的临床试验,也需要专业的人来进行,比如现在就有专门做这样业务的CRO公司。
所以,国家、社会、医院对临床试验的支持,应该思考如何有效地将医生和临床CRO公司进行组合,以获得更高质量的临床试验结果。这应该是一个趋势,但是如何监管、如何保证透明化,也需要智慧。
另一方面,还是应该鼓励一线医生参与临床研究,如果是研究者发起的临床研究,在不影响重点临床试验的情况下,入组试验的人数合理即可。这种试验只是早期的试验,一般不可能获得严谨的结论,但是可以对临床效果做出一定的评判,成为展开大规模临床试验的依据。
中国开展的瑞德西韦等药物的临床试验,之所以无法对入组人数做出比较准确的判断,一个重要的原因是之前没有早期试验的结果来作为依据。而也正是在基于中国临床试验经验的基础上,国外的临床研究才及时调整了入组人数,保证试验能够获得一个阳性的结果。
如今疫情在中国已经得到非常有效的控制,但是并不能说疫情就已经完全结束。目前在局部地区,还是有本土病例发生。同时,因为全球广泛发生的疫情,输入病例也是中国面临的压力。
面对这些病例,中国还是有机会继续一些临床研究。没有了突如其来的疫情压力,希望中国能够从容进行更新的具有前景的临床试验,珍惜宝贵的机会。
参考文献
1. 陈军, et al., 洛匹那韦利托那韦和阿比多尔用于治疗新型冠状病毒肺炎的有效性研究. 中华传染病杂志, 2020. 38(00): p. E008-E008.
2.Cao, B., et al., A Trial of Lopinavir-Ritonavir in Adults Hospitalizedwith Severe Covid-19. N Engl J Med, 2020. 382(19): p. 1787-1799.
3.Hung, I.F.-N., etal., Triple combination of interferonbeta-1b, lopinavir–ritonavir, and ribavirin in the treatment ofpatients admitted to hospital with COVID-19: an open-label, randomised, phase 2trial. The Lancet.
制版编辑 | 皮皮鱼

0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号