阅读:0
听报道
撰文|越君
责编|李娟
● ● ●
还记得几年前火遍全球社交网络的“冰桶挑战”吗?那次活动让人们了解到一种神经病变,即“渐冻人症”。与广为人知的老年痴呆症和帕金森病一样,这类疾病都属于神经退行性疾病(Neurodegenerative Disorders),是由于大脑和脊髓中的神经细胞功能衰退所致。因为人体的所有运动,包括说话、吞咽以及呼吸肌的活动等,都是由运动神经细胞支配,一旦神经细胞失去正常功能,带来的就是这类足以让人崩溃的疾病。
舞蹈症的独特之处
在神经退行性疾病中,还有一种疾病格外引起科学家的注意——亨廷顿病(Huntingtin Disease),又称舞蹈症,因发病者肢体像跳舞般不受控运动而得名。亨廷顿病与其他同类疾病有个共同点,都是由致病蛋白质在神经细胞里的异常积累所引起。比如,亨廷顿病的致病蛋白是由变异HTT基因编码的HTT蛋白。该疾病之所以让很多科学家颇感兴趣,还因为亨廷顿病还有其独特之处。
首先,亨廷顿病是遗传病,单一基因HTT的突变就足以引发该病。这种单基因遗传的性质有利于对其致病机理的探索,基于此,科学家建立了相对可靠的疾病遗传学模型,进而严密地证明了HTT致病蛋白积累与该病的因果关联。
其次,致病基因HTT的突变很特别。与单个核苷酸突变引起的遗传病不同,HTT基因突变是一段三核苷酸重复序列的重复数改变。具体来说,该基因有一段CAG重复序列,正常人中CAG的数目小于36个,而大于等于36个就会导致疾病。CAG序列编码的氨基酸是谷氨酰胺(Glutamine),简写是Q,因此CAG重复序列对应的氨基酸是QQQ…Q,常用polyQ来表示。亨廷顿病人的polyQ过长,大多数病人有40多个重复,使得HTT蛋白成了导致神经细胞死亡以及发病的“毒蛋白”[1]。
这里有一个非常重要又有趣的问题,HTT蛋白本来不具有毒性,甚至是胚胎发育所必须的蛋白[2],为什么该蛋白的polyQ长度超过一定阈值就产生毒性,导致如此严重的疾病呢?
毒性从哪里来
科学家提出了两个模型尝试对此问题作出解释。
第一个模型被称为线性格栅模型(Linear Lattice Model)。科学家认为,polyQ长链好似铺有整齐枕木的铁轨,由相同的结构单元组成,“枕木”之间的结构单元可能本身就自带毒性(intrinsictoxicity),polyQ越长,蛋白的细胞毒性越强,达到一定程度就会造成疾病。简单来说,即polyQ中具有线性排列的含有内在毒性的结构单元。
有哪些证据支持该模型呢?首先,体外生化实验和结构生物学的研究发现,polyQ的抗体可以与很短的polyQ片段(10Q)结合[3,4],而不同polyQ抗体与野生型短polyQ以及突变型长polyQ的结合模式相同[3,4]。这提示polyQ长链是由相同的结构单元组成。其次,有关蛋白功能的研究显示,病人的发病年龄与polyQ长度有关,polyQ越长,病人的发病年龄总体来说越早[1]。另有研究表明,如果把小鼠Htt基因的7个CAG重复完全删除,小鼠不但没有出现缺陷,反而延缓了衰老,变得更健康了[5]。
然而,该模型仍然很难解释为何增长的polyQ会突然产生严重毒性,较短的polyQ却完全正常。而且,相关体外实验也无法反映细胞中的情况,因为细胞内的polyQ可能具有多种不同构象。
于是,有科学家提出了第二个可能的解释——毒性浮现构象模型(Emergent Conformation),即变异蛋白的polyQ长度达到一定阈值之后,会出现与“整齐的枕木”不同的新的构象单元,而这种新的构象单元是导致神经毒性的主要原因。这种解释并没有否定线性格栅模型,只是推测除了线性格栅构象之外,polyQ长链上还出现了真正具有毒性的“浮现构象”,因此是多种构象的混合(图1,摘自参考文献[6])。
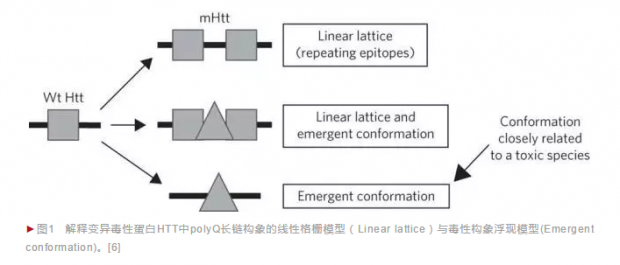
毒性浮现模型的支持证据相对来说非常间接,主要是基于不同polyQ抗体识别的变异HTT蛋白信号与细胞毒性的相关性不同,因此推测变异HTT蛋白过长的polyQ具有多种构象,其中包括了最具毒性的“浮现构象”[6]。这里相关性的计算基于很多非直观的假设,缺乏更直接的证据,被不少研究反对[4,7]。
由此可以看出,以上两种观点最大的争议在于,变异蛋白中过长的polyQ是否存在多种不同的构象,也就是构象是否存在“多态性”。
这可能是生物化学的一个根本问题。因为我们知道,蛋白质的三维结构由该蛋白质的氨基酸序列所决定,这被称为Anfinsen法则(Anfinsen’s Dogma),并获得1972年的诺贝尔奖。该法则也是结构生物学的基础,因为结构生物学的研究基于这样的假设:蛋白质在特定体外条件下的三维结构与细胞内的三维结构相同。
然而,有不少蛋白会“错误折叠”(misfolding)并产生不同的构象。在神经退行性疾病中,“错误折叠”的蛋白容易聚集在一起并最终形成蛋白沉淀。那么,可溶性蛋白质的单体是否也会违背Anfinsen法则,通过“错误折叠”产生多种不同的构象,进而产生不同的毒性,最终导致疾病呢?目前还没有答案。而且,如果真的存在“多态性”,为什么其中某些构象的毒性会更高?这也是关于疾病致病机制的根本性问题。
但是研究上述问题很难。变异HTT蛋白非常大(>300kDa),结构难于解析。即使可以解析,目前结构生物学的手段无法在细胞非变性条件下解析特定目标蛋白的精细结构,而体外纯化的条件下则可能无法获得蛋白折叠的各种构象,更加无法进一步证明各种构象可能的功能(毒性)差异。
用“蛋白质降解”来探寻答案
2017年9月4日,Nature Chemical Biology在线发表了一篇论文[8],由复旦大学鲁伯埙课题组完成,该论文从一个崭新的角度研究上述问题:蛋白质降解。
细胞中,新蛋白质在不断地合成,完成功能的或者“错误折叠”的蛋白质则被不断地降解,生成小的肽段或氨基酸。主要有两个可能的因素影响蛋白降解速率:蛋白构象和蛋白功能。
一方面,蛋白构象很可能影响其降解速率,如果同类细胞的同一蛋白有着不同的降解速率,意味着该蛋白存在不同的构象;另一方面,蛋白降解是蛋白功能的终极调控。已有数据表明,疾病蛋白的降解速率与其神经毒性呈非常显著的负相关,即致病蛋白降解越慢,其毒性越大[9]。因此,了解蛋白本身是否具有构象的“多态性”,以及不同构象蛋白的毒性是否具有“多态性”,都可以从研究蛋白的降解速率入手。如果不同polyQ抗体识别的HTT蛋白的降解速率不同,则证明的确存在不同构象,而且降解速率较慢的构象的毒性可能较大。
为了检测以上假说,鲁伯埙课题组测得了同一组样品中被不同polyQ抗体(主要有MW1和3B5H10两种)所识别的HTT蛋白的降解速率。他们所使用的测量方法被称为CH-chase,是基于点击化学(Click Chemistry)和均相时间分辨荧光(HTRF)技术而建立的。
首先,他们选用的细胞模型是病人的真皮成纤维细胞,这是因为该细胞中有全长的变异HTT蛋白,并且检测不到寡聚体或多聚体,排除了可能的干扰因素,进而只检测可溶性HTT蛋白单体的降解。研究显示,在三种不同的病人真皮成纤维细胞中,polyQ抗体3B5H10所识别的HTT的降解速率显著低于另一种polyQ抗体(MW1)所识别的HTT的降解速率,从而直接证明了不同polyQ构象的存在。这种降解速率差异在过表达的体系中一样可以被检测到。有趣的是,这种差异只在过长的polyQ(46Q,72Q,100Q)中存在,在野生型长度的polyQ(18Q及23Q)中并没有检测到,从而证明了过长的polyQ出现了“浮现构象”,因此产生了“多态性”。
那么,是什么因素减慢了这类HTT蛋白的降解速率呢?鲁伯埙课题组的进一步研究发现,导致3B5H10抗体所识别的HTT蛋白的降解显著变慢的原因,是因为其能够抵抗自噬作用。
自噬(Autophagy)是细胞清除蛋白的重要手段,自噬小泡包裹住蛋白,并将其运送至溶酶体完成降解。自噬分子机制的发现获得了2016年的诺贝尔奖。传统观点认为,自噬作用是非选择性地,能随机清除任何蛋白。然而近年研究发现,有些自噬过程中,自噬小泡会利用自噬受体蛋白p62或NBR1等有选择性地吞噬特定的蛋白,这个过程称为选择性自噬。
鲁伯埙课题组的研究发现,在病人细胞、小鼠大脑组织乃至病人大脑组织中,变异HTT蛋白的赖氨酸63型泛素化几乎缺失,因此无法被选择性自噬受体蛋白p62所识别,也就无法被选择性自噬降解。这就解释了为何这种HTT构象降解速度显著变慢(图2)。
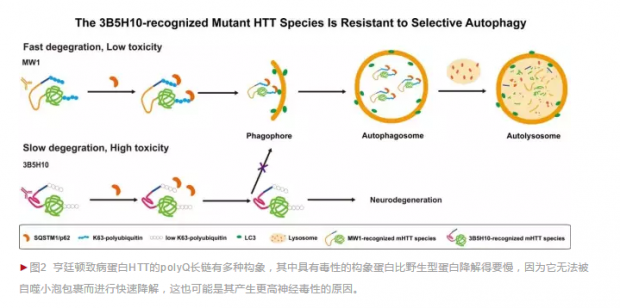
这项最新研究揭示了导致亨廷顿疾病的变异蛋白HTT的polyQ长链存在多种不同的构象,并因此导致了不同的降解速率,产生了不同的毒性。这种“多态性”可能补充了对“Anfinsen法则”的认识。同时,该研究进一步明确了选择性自噬作用的机制,发现选择性自噬比之前认为的要更具选择性:它不仅能识别特定的蛋白进行降解,甚至能识别同一蛋白的不同构象。
该研究在疾病研究领域也有重要意义,不仅刷新了科学家对疾病机制的认识,而且提供了治疗疾病的新思路。例如,可寻找化合物迫使疾病蛋白从慢速降解的构象转变为快速降解的构象,达到治疗疾病的目的。不过,仍然有几个关键问题亟待解答,比如,为何polyQ构象的改变会影响HTT的泛素化以及选择性自噬的识别,其他polyQ蛋白是否具有类似的性质等,值得后续研究。
参考文献
1.Walker, F.O., Huntington's disease. Lancet, 2007. 369(9557): p. 218-28.
2.Nasir, J., et al., Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell, 1995. 81(5): p. 811-23.
, P., et al., The structure of a polyQ-anti-polyQ complex reveals binding according to a linear lattice model. Nat Struct Mol Biol, 2007. 14(5): p. 381-7.
4.Klein, F.A., et al., Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum Mol Genet, 2013. 22(20): p. 4215-23.
5.Zheng, S., et al., Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet, 2010. 6(2): p. e1000838.
6.Miller, J., et al., Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol, 2011. 7(12): p. 925-34.
7.Owens, G.E., et al., Anti-PolyQ Antibodies Recognize a Short PolyQ Stretch in Both Normal and Mutant Huntingtin Exon 1. J Mol Biol, 2015. 427(15): p. 2507-19.
8.Fu, Y.W., P. Pan, Y. Sun, X, Yang, H. Difiglia, M. Lu, B., A toxic mutant huntingtin species is resistant to selective autophagy. Nature Chemical Biology, 2017.
9.Tsvetkov, A.S., et al., Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol, 2013. 9(9): p. 586-92.

话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}