阅读:0
听报道

撰文|吴培煌
责编|李 娟
● ● ●
癌症是威胁人类生命健康的重大疾病之一。多年来,科研人员一直致力于研究开发治疗癌症的药物,而发现具有普遍适用性的药物是医学界孜孜以求的目标。癌症研究权威期刊Cancer Cell在创刊10周年之际,回顾了过去10年中人类的“抗癌”征途——理想癌症治疗效果应该是特异性杀伤癌细胞、预防耐药性发生,以及降低毒副作用。根据患者的个体信息制定治疗方案,从分子水平差异控制治疗效果或毒副作用等,并根据患者的临床反应适时调整治疗措施,以实现癌症治疗“私人订制(精准)”的目标。
那么,从“普适性”到“私人定制(精准)”,癌症治疗模式的突破是如何发生的呢?
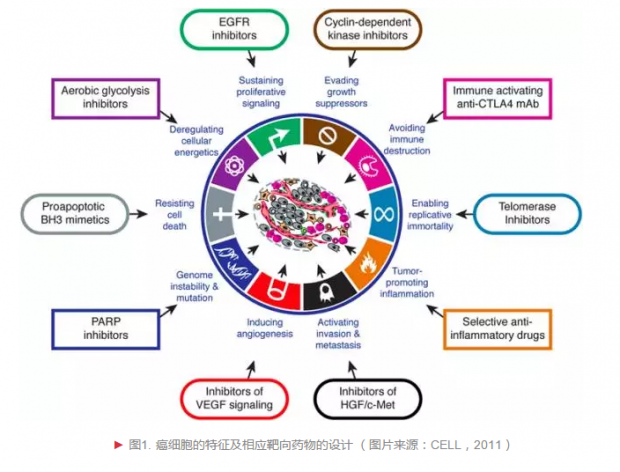
首先,我们要了解治疗对象的特征,才能做到精准。目前大多数观点认为,癌症是基因突变导致,而非入侵机体的异类,基因结构和功能发生变化使得细胞发生癌变。癌细胞能够无限增殖,抵抗细胞死亡,消耗并占用大量营养资源。有些癌细胞能量异常,具有生成血管的能力。有些癌细胞能向其他组织器官浸润和转移。有些癌细胞呈现异质性,不断改变自己的“面貌”以逃脱免疫系统的巡逻和监视——这些是癌症细胞有别于正常组织细胞的特征(图1),也是研究人员开展癌症治疗研究的基础[1]。
化疗是癌症的传统治疗手段,化疗药物可以随着血液循环到达全身各处,消灭转移和扩散的癌细胞。这种治疗方式具有普遍适用性。癌细胞再生需要各种蛋白的合成,而化疗药物正是针对癌细胞具有活跃的增殖特性来杀伤癌细胞。它通过各种手段干扰DNA,防止细胞再生所必需的蛋白质合成。但是,机体内存在一部分代谢旺盛的正常血液细胞,淋巴细胞,表皮和胃肠道粘膜细胞等,因为它们活跃的再生能力,往往会遭到化疗药物的攻击,导致药物副作用的发生。而且,某些癌细胞分泌产生的细胞膜蛋白(P-糖蛋白)能够将化疗药物排出细胞外,或对化疗药物进行充分分解稀释,使细胞内的药物浓度不足以杀死癌细胞,使化疗药失去原有细胞毒作用,癌细胞产生抗药性,导致治疗失败。
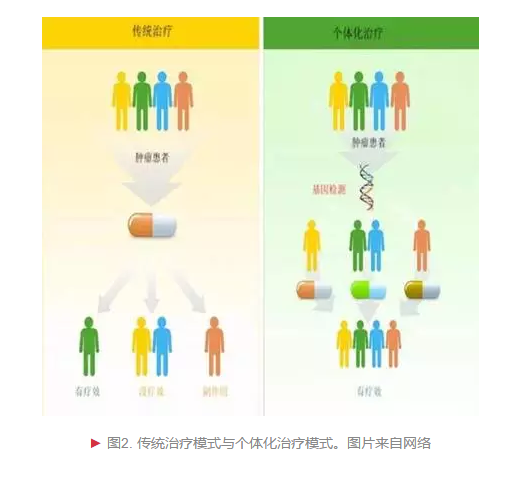
近年来,随着细胞分子生物学研究领域的不断发展,以及相关临床实践经验的累积,治疗癌症的新思路层出不穷。科学家们采用不同新技术,对患者的癌细胞进行基因层面分析,以极其精确的方式定位细胞的位置、组成和功能,可以更好地去瞄准目标,杀伤癌细胞。个体化癌症治疗方案的核心是合理设计药物。分子靶向药物“甲磺酸伊马替尼”(商品名:格列卫)的研发进程就体现了癌症治疗模式从普适到精准的突破[2]。
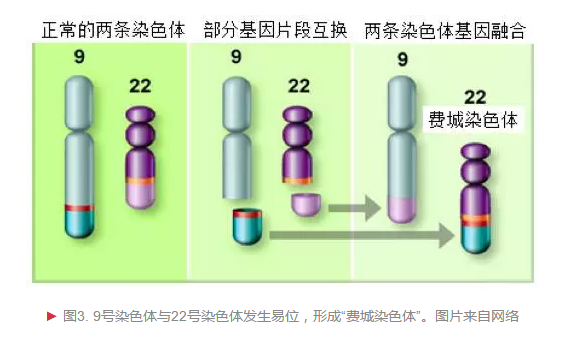
慢性粒细胞白血病(chronic myeloid leukemia, CML,简称慢粒)是一种造血干细胞恶性克隆疾病,现在我们知道,对比其他恶性肿瘤,它的致病机理并不复杂。1960年,Peter Nowell发现这种癌细胞存在染色体异常;1973年Janet Rowley通过染色体染色技术确定费城染色体为9号染色体与22号染色体部分区域位置交换;后来随着癌基因研究的兴起,Annelies de Klein证实费城染色体是9号染色体上原癌基因ABL和22号染色体上的BCR(染色体易断裂区)基因交换融合(图3)而成。
蛋白质的合成由一整套密码控制,密码信息就存在我们的染色体DNA中。在编码合成蛋白的过程中,密码的解读需由阅读框进行,在编码过程中是没有分隔的,三个连续的碱基为一个密码子,翻译为一个特定的蛋白质氨基酸序列。密码阅读是从一个固定的起点开始,这就好像我们跑步时有起跑线和终点线,有特定的密码(碱基序列)作为起始信号和终止信号。细胞的整套密码分布在各条染色体上,各自负责特定蛋白的翻译。慢粒中基因融合的费城染色体,导致细胞里原先那套标准遗传密码发生变化,ABL密码连接了BCR密码就翻译出了新的蛋白——酪氨酸激酶BCR-ABL,这种酪氨酸激酶活性持续升高就导致了慢性粒细胞不断增殖[3, 4]。
知道了酪氨酸激酶活性升高是罪魁祸首,能不能设计药物抑制这个融合蛋白(酪氨酸激酶BCR-ABL)的活性,从而达到治疗慢性粒细胞白血病的目的呢?这个想法首先来自肿瘤学家Brian Druker,1993年,他开始与诺华公司生化学家Nicholas Lydon合作,最终针对费城染色体融合基因BCR-ABL设计合成出了靶向药物[5],经过一系列细胞学实验、动物实验到临床试验各角度筛选并评估药物安全性和有效性[6]。生物物理学家John Kuriyan则阐明了药物与BCR-ABL作用的生物物理学机制。在此基础上,科学家最终研发得到了被称为“格列卫”的药物。
“格列卫”治疗慢性粒细胞白血病取得了巨大成功,但是,癌细胞耐药性的产生引起了人们极大的关注。Druker对“格列卫”的治疗进行了5年随访,发现大约35%的患者出现耐药或药物副作用。随后,Tomi Sawyer发现“格列卫”的治疗过程中,BCR-ABL的基因位点发生突变,而突变后产生的BCR-ABL融合蛋白的空间构象发生了改变,导致“格列卫”无法与之结合。因此,Sawyer联合百时美施贵宝制药公司合作研发新的“格列卫”类似物,使新药物可以同时作用于突变和未突变的BCR-ABL蛋白,可以用于对“格列卫”耐药或不能耐受的费城染色体阳性的慢粒患者[7]。至此,酪氨酸激酶抑制剂“甲磺酸伊马替尼”的问世实现了慢粒治疗史上的飞跃,达到了延长患者生存期的目的[8]。
基于对癌症治疗方式的深入研究,我们知道要找到一种广谱治疗癌症的灵丹妙药基本是不可能的。“格列卫”在治疗慢性粒细胞白血病上的巨大成功,预示癌症治疗将进入一个“精准”时代。癌症治疗方式的突破有赖于对癌症的基础研究、新技术更新、药物设计研发及临床数据的深入总结,同时,也依赖多个国家几代药物学家、肿瘤学科研人员和临床医生的集体智慧,更是基础学科、药物研发公司和临床医疗中心密切合作的转化医学经典。
参考文献:
1.Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144(5):646-674.
2.Longo DL: Imatinib Changed Everything. The New England journal of medicine 2017, 376(10):982-983.
3.Q. G, DALEY RA, VAN ErrEN, BALTIMORE D: Induction of chronic myelogenous leukemia in mice by the P210bcr abl gene of the Philadelphia chromosome. Science 1990, 247:824 -830.
4.Nowell PC: Discovery of the Philadelphia chromosome: a personal perspective. The Journal of Clinical Investigation 2007, 117(8):2033-2035.
5.Elisabeth Buchdunger, Jürg Zimmermann, Helmut Mett, Thomas Meyer, Marcel Muller, J. B, Druker, Lydon NB: Inhibition of the Abl Protein-Tyrosine Kinase in Vitro and in Vivo by a 2-Phenylaminopyrimidine Derivative. Cancer Research 1996, 56:100-104.
6.Druker BJ TM, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL.: Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine 2001.
7.Thomas O’Hare RP, Eric P. Stoffregen, Jeffrey A. Keats, Omar M. Abdullah, Erika M. Moseson, Victor M. Rivera, Hao Tang,, Chester A. Metcalf III RSB, YihanWang, Raji Sundaramoorthi, William C. Shakespeare, David Dalgarno,, Tim Clackson TKS, MichaelW. Deininger, and Brian J. Druker: Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potentATP-based oncogenic protein kinase inhibitor: implications forCML. BLOOD 2004, 104(8).
8.Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. The new England of journal of medicine 2017.

话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}