编者按:很多人可能对Mark Waller教授比较陌生,对他任职的单位上海大学也不熟悉,但最近Waller在Nature杂志上发表了一项人工智能领域的重要研究,化学界的“AlphaGo”由此诞生,引起国内外制药领域高度关注。这可能也是上海大学近年为数不多能登上《自然》的研究,Waller为上大物理系教授,2016年3月全职加入上海大学。为此,我们邀请同济大学生物信息学教授、IEEE会员刘琦对这项研究进行深入的解读,以及剖析化学界“AlphaGo”仍存在的短板。刘琦教授目前主要致力于计算机和生命科学的智能计算和机器学习的交叉研究。

2018年3月29日,国际顶尖学术期刊Nature在线发表了上海大学教授Mark P. Waller团队利用深度学习的逆向合成路线设计药物的研究论文(Planning chemical syntheses with deep neuralnetworks and symbolic AI)。
我和Mark曾在药明康德的小型会议上有过交流,知道他的工作早在一年前已发布在预印本arXiv上,现在终于正式发表,表示祝贺。该工作是Nature杂志继2016年发表的基于机器学习技术进行晶体制备策略预测的封面文章之后[1],再次发表的一项基于人工智能技术进行药物自动化开发的研究成果。基于AI加速药物研发,正越来越受到业内广泛的关注。
相比于《自然》杂志2016年发表的晶体合成反应预测工作,本次工作关注于计算机辅助的合成设计(Computer-assisted synthesis planning,CASP),其核心又被称为反合成分析(Retrosynthesis analysis)。
反合成分析可认为是小分子正向反应预测(Forward reaction prediction)的逆过程。有机合成大师埃里亚斯·科里(Elias Corey)于20世纪60年代首次提出反合成设计的思想,并据此设计了第一个初步实现CASP的软件LHASA。1990年,科里教授由此获得了诺贝尔化学奖。
实现反合成分析有两个关键点:1)对有机化学反应的深刻了解,即规则的掌握;2)基于这些规则,对目标分子进行递归式拆分,最终获得易得原料,即路线的设计。传统的CASP通常是基于规则的专家系统,这些规则可人为地进行编码或者从数据库中用算法进行自动提取,而路线的设计则主要依赖于启发式算法评估拆分的合理性。传统反合成分析方法存在着诸多问题制约其广泛应用,包括泛化能力差、缺乏化学合理性等,常令合成化学家望而却步。
在这项研究工作中,Waller团队将深度神经网络及强化学习等概念整合至一个通用的架构中,提出了采用三种不同的神经网络结合蒙特卡洛树搜索(Three different neural networks together with MCTS)的算法框架(3N-MCTS)。这三种神经网络分别为:拓展策略网络(Expansion policy network)——用来搜索当前位置可能存在的单步逆向化学变换路径(Transformation);筛选网络(Filter network)——用来对反应的可行性做出判断;展示策略网络(Rollout policy network)——用来在展示步骤中应用多次采样方法对搜索节点进行定量评价。该工作相对于传统基于规则的反合成设计来说,大量借鉴了深度神经网络及强化学习的思想,是对传统方法的一次重要的改进,这也是该工作能够发表于Nature的原因之一。
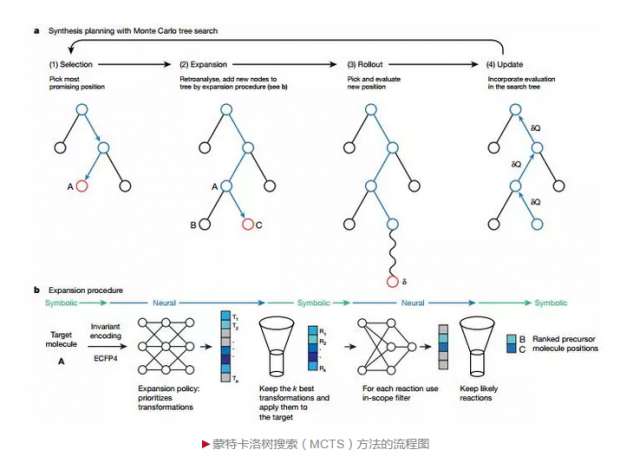
蒙特卡洛树搜索(MCTS)是逆合成路线规划的核心框架,分成四个步骤:选择(Selection)、拓展(Expansion)、展示(Rollout)、更新(Update)。其中:拓展策略网络(Expansion policy network)与筛选网络(Filternetwork)组成一个流水线(Pipeline),应用在拓展步骤(Expansion);展示策略网络(Rollout policy network)则应用在展示步骤(Rollout)。这四个步骤中,选择和拓展步骤非常容易理解,即首先选择当前最适合拓展(位置权重最大)的化合物节点,进行可能的合成策略预测。其中拓展策略网络是基于深度神经网络进行合成策略预测,筛选网络是将前一步预测的合成策略进一步进行可行性判定。展示和更新步骤的核心思想为马尔可夫决策过程(Markov decision process),我们可将该过程类比于机器下棋,机器在展示步骤并不做真正的分支延展,而是通过反复的采样对局来探索可能的状态空间。
所不同的是,在逆合成分析中,机器并没有对手,其探索结束的标准也不是输赢,而是探索所获得的所有合成原料均可归属于事先定义好的一个易得原料集合,在文中被称之为All molecules are solved,或探索达到了事先定义的树最大深度值。其展示的过程将依次从展示策略网络(Rollout policy network,类似于Expansion policy network,但是网络结构更加简单,方便快速进行rollout)预测的rollout rules中进行选择,同时探索结束后将根据不同的探索结果获得不同的反馈(强化学习)。在更新步骤中,将根据展示步骤的多次模拟,进行通过节点的访问次数计数以及位置权重更新。直观来说,模拟过程中访问次数越多的节点,其位置权重越大,那么在下一次探索中也就更可能被访问,这个过程可以用一句俗语总结: “世上本无路,走的人多了,也就成了路!”,这也是MCTS核心思想的一种体现。
如果读者对于DeepMind的AlphaGO有所了解的话,一定会惊奇的发现,该工作的四个步骤,可类比于AlphaGO一文中所提出的四个步骤(Selection, Expansion, Evaluation, Backup)。2016年,DeepMind团队在Nature杂志发表AlphaGO研究成果[2],其升级版本AlphaGOZero以封面文章形式于2017年在Nature杂志发表[3]。顶级杂志连续三年发表四篇AI的应用,并且两次均为封面文章,说明AI技术的应用受到了极大的关注。
3N-MCTS的验证以2015年之前发布的化学反应作为训练数据,以2015年之后发布的化学反应作为测试数据。在双盲AB测试中,来自中国和德国的45位有机化学家对9个不同目标分子的文献报道的合成路线和3N-MCTS生成的合成路线进行判断,有57%的化学家选择3N-MCTS生成的路线,43%的化学家选择了文献报道路线,并且双方没有选择上的统计显著性差异,表明3N-MCTS生成合成路线的水平与文献报道路线水平相当。同时在对于3N-MCTS生成的合成路线和广度优先搜索(heuristic BFS)生成的合成路线的比较中,受试科学家显著倾向于选择前者,表明3N-MCTS生成合成路线的水平显著优于BFS的基线水平。
最后,Waller团队进一步论证了上述四个步骤对反合成分析整体性能的提升均有贡献,这一点上,大家也可以类比于AlphaGO一文进行理解。
此次,Waller团队的研究成果是当前化学合成领域的一个重要的突破,对于化合物合成特别是药物合成具有重要的意义。目前,计算机辅助化合物逆向合成主要有两种解决方案:1)基于人工规则的逆向合成路线设计,这种方案目前的典型代表为韩国蔚山国立科学技术研究所(UNIST)Bartosz Grzybowski教授团队开发的Chematica系统,该系统遵循50000条有机合成规则并基于这些合成规则进行合成路线的设计;2)基于深度学习的逆向合成路线设计,Waller团队的3N-MCTS算法框架即为该方案的典型代表。
纵观3N-MCTS的整个设计方案,和AlphaGo的整体思路非常类似,均采用了深度神经网络+蒙特卡洛树的实现方式,将合成路径的选择类比于围棋对弈中落子路径的选择。这种方法值得肯定,也取得了令人满意的结果。最后我对该工作做一个总结和展望:
首先,该工作能发表在Nature杂志,可以说既是“意外”, 又“惊喜”。“意外”系指其整个设计框架借鉴于AlphaGO, 故可能从方法学角度来说并未做实质性的创新;“惊喜”系指该工作做了一个非常巧妙的类比和应用,尝试解决化学合成领域的一个重要的问题,并取得了理想的结果,对该领域产生了重要的贡献。
其次,3N-MCTS的整个方案基于深度神经网络构建,故需要大量的高质量标记样本进行训练。而化学合成反应的标记样本并不容易获得。该工作所用的训练样本均来自于Reaxys数据库,该数据库是商业数据库,不可公开获取。进一步扩充相关的化学反应标记数据,将能够获得更好的预测效果。
再者,前述所说的筛选网络是一个二分类的深度神经网络,它基于数据库中的阳性数据(可行路线)和阴性数据(不可行路线)进行训练。而数据库中的阳性数据远多于阴性数据。为了解决这个问题, Waller团队采用了data sampling 和data augmentation的方法进行负样本生成,但并不能够穷尽所有的阴性数据,并且阴性样本的选择和生成将对整个系统的预测性能产生重要的影响, 这一点值得未来进行系统的讨论。
最后,目前其输入化合物预测的编码均采用基于Smile格式的ECFP4指纹编码,该指纹编码维度很高,易造成过拟合,未来的方向可以考虑进一步尝试其他的化合物编码方式进行预测。Waller团队也提到其目前的模型基于已有的训练数据,对某些特定反应机理、三维结构或互变异构体对复杂化合物合成的影响还无法考虑,在复杂天然产物的逆合成以及立体化学选择性预测方面有很大改进空间。同时,目前的方法只提供了可能的设计路线,而对反应的条件还无法做出准确的预测。
总体来说, Waller团队的工作是基于AI对于药物逆合成路线设计的一次有益尝试,同时也存在诸多改进空间。另外,我们注意到该文的第二作者Mike Preuss来自于英国BenevolentAI公司,该公司目前是也已基于AI进行药物研发的独角兽公司, 说明学术界和工业界均对于AI加速新药研发产生了关注。我们期待未来AI技术在该领域再次获得激动人心的研究成果。
相关文章
[1] Raccuglia P et al. Machine-learning-assistedmaterials discovery using failed experiments, Nature, 2016.
[2] Siliver D et al. Mastering the game of Go withdeep neural networks and tree search, Nature, 2016.
[3] Siliver D et al . Mastering the game of Go without human knowledge,Nature, 2017.


0
推荐
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}