阅读:0
听报道

2020年11月30日,谷歌旗下DeepMind公司研发的人工智能系统AlphaFold在第14届国际蛋白质结构预测竞赛(CASP)中摘得桂冠。
当日,DeepMind在官网发布新闻稿,称对于困扰生物学界50年的 “蛋白质折叠” 难题,AlphaFold系统已成为业内专家认可的解决方案;Nature新闻更是以 “it wil change everything”(将改变一切)作为标题,指出DeepMind在解决蛋白结构问题上“迈出一大步”。
这些激动人心的表述,让网友们惊呼:如果机器预测蛋白结构这么准,结构生物学家们的饭碗,要被机器抢走了吗?
撰文 | 戴 威
责编 | 陈晓雪
1 AlphaFold预测结构夺冠,再次名声大噪
了解一种新的蛋白结构有两条路,一是靠实验去 “看”,二是根据氨基酸序列用计算机去 “算”。
可以说,AlphaFold是目前 “算” 这条路上的冠军。
利用计算预测蛋白质结构的CASP竞赛创始于1994年,如今已成为评估预测技术的黄金标准,也是这一领域交流新技术的国际平台。该竞赛以全局距离测试(Global Distance Test,GDT)计分,满分100分,分数越高,意味着每个氨基酸残基(蛋白链上的小球)离实验测得的正确位置越接近。用于竞赛的蛋白质均为新近实验破解的、且未公开发表的研究。
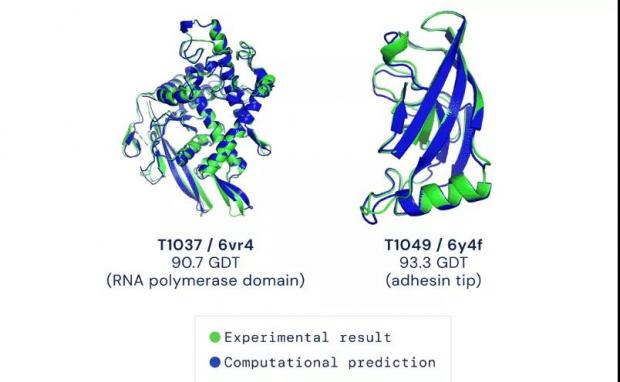
自由建模环节中AlphaFold的模型与实验数据高度贴合。蓝色为计算预测,绿色为实验结果。图源:
在今年CASP比赛中,组织方在5月到8月间放出氨基酸序列,上百个参赛团队可在5月至9月中旬间提交模型。
最终,DeepMind 旗下AlphaFold系统拿下中位数92.4GDT的高分,就是在难度较高的自由建模环节也达到了87分的中位数,比第二名高出了25分。
马里兰大学教授、竞赛创始人之一的约翰·蒙特(John Moult)教授认为,90 GDT左右的分数可认为与实验方法获得的结果相竞争。
“这绝对是惊人的突破。对于超大分子量的单一蛋白,体外表达上就存在困难,更别提更大的复合物,所以在获得高质量的样品用于结构解析是具有挑战性,以及费时费力费钱。能够利用准确预测的结构进行分析,对于相应的研究意义非凡。” 对于AlphaFold的成绩,清华大学生命科学学院研究员李赛不吝赞美。结构生物学家李赛的研究方向是用冷冻电镜解析生物样本,而清华大学拥有国际一流的冷冻电镜平台。
AlphaFold预测的效果如此之好,与实验结果只有小幅差异,甚至让人怀疑差异是否源于实验结果不够精细。“跟实验方法对比的话,有时候说,分辨率如此之高,有时候并不知道这个误差是来自于结构的预测,还是来自于实验方法分辨率的限制。所以这个消息是让人很振奋的。” 李赛告诉《知识分子》。
蛋白质由长长的氨基酸链组成,而仅有正确的氨基酸是不够的,这些链条必须扭卷成特定的三维结构,蛋白质才有活性,这一物理过程被称为蛋白质折叠。氨基酸链未折叠或折叠错误的蛋白质,都不能正常发挥生理功能。因此,探究蛋白质的结构对于研究其功能十分重要。
1972年,美国生物化学家克里斯迪安·安芬森(Christian Boehmer Anfinsen)提出假设,氨基酸序列能完全决定蛋白质结构。但氨基酸序列折叠的可能性多到数以亿计,仅通过氨基酸序列,并不能得到完整的蛋白质结构,因此在过去几十年里,计算预测只是实验手段的辅助。
解析蛋白质结构,现有的常用实验方法有三种:核磁共振、X射线晶体学和冷冻电镜。
如今,已有约17万蛋白质的结构经实验破解,并上传至蛋白质数据银行(Protein Data Bank,PDB)公开。
随着海量的序列和结构数据积累,预测结构不再是根据氨基酸序列“空算”,而有了学习的依据。利用这17万公开的蛋白质序列和结构数据,以及已知序列而未知结构的蛋白质数据库,DeepMind 对 Alphafold 进行训练。
2018年,DeepMind 推出 AlphaFold1 参加第13届CASP竞赛,今年的系统则是新版本AlphaFold2,使用了大约128个TPUv3核(相当于100-200个gpu)进行数周运算,算力更强。
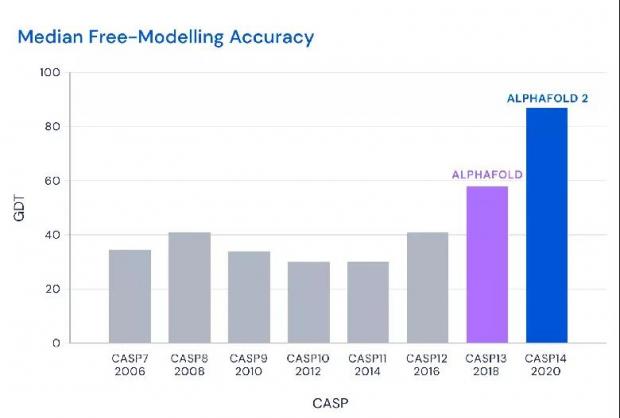
相比于上一代AlphaFold,新版本在中位数准确性上更进一步。图源:
2 结构生物学家:AlphaFold目前适用于理想环境的小型结构
李赛表示,新闻中的AlphaFold建模的对象都是一些分子量较小的或较基础的结构。
从简单到复杂、分子量从低到多,蛋白质结构可分为四级。其中,一级结构指线性的氨基酸序列;二级是形成稳定结构的氨基酸链,比如线圈状的α螺旋、锯齿状的β折叠;三级是几个二级结构形成的三维结构;四级结构是蛋白质复合物。
目前DeepMind展示的建模模型处于二级结构到三级结构之间。
展示的可能是(分子量)比较小的,可能是由几个α螺旋或者几个β折叠这样的二级结构,或者是一些结构域(超二级结构)、小蛋白质结构。” 李赛解释,蛋白越大、折叠的不确定性就越大。对于蛋白中稳定的结构域,通过氨基酸序列就可预测二级结构。但随着结构尺度“升级”,会出现一些折叠不确定的部分,“这些对于预测是蛮难的,即使是实验方法都不能保证测出来。
清华大学生命科学院院长王宏伟表示,“AlphaFold目前还主要是预测单链蛋白或结构域,无法预测较大的蛋白质复合体,尤其是包含很多不同组分的生物大分子机器的结构。” 他解释说,这主要是因为蛋白和蛋白的相互作用非常复杂,存在极多的可能性,即使实验手段冰山一角也只揭示出冰山一角。
对于复杂的蛋白质或蛋白复合物,科学家们现在主要通过冷冻电镜来解析。在核磁共振、X射线晶体学、冷冻电镜三种方法里,最初用于核物理的核磁共振要求生物样本分子量最小(15~25Kd),分辨率最高,达1埃以下;X射线衍射法对样本大小(不超过200Kd)的要求和分辨率都居中,但样本必须结晶;冷冻电镜的分辨率原不如前两者,适用于研究单个较大的生物分子,但2013年后技术革命,分辨率已达到原子级。
李赛告诉《知识分子》,结构生物学和冷冻电镜方法现在的研究重点依然是蛋白质三维结构,包括蛋白蛋白复合物、蛋白核酸复合物、糖蛋白等等。
除了可能的蛋白大小和结构复杂度的区别,李赛认为人工智能预测蛋白结构更依赖于比较理想化的环境。
而理想状态与蛋白质的自然状态是不同的。
李赛解释说,“结构生物学发展的大的趋势是做原位蛋白结构,这个就更复杂了。比如说一个病毒,(想研究)嵌在膜上的蛋白与另外一个蛋白的关系,要把结构解析出来。除非计算的方法能在哪一天模拟整个病毒、细菌、细胞,并把所有复杂因素都考虑,不然无法做到接近自然。”
3 机器预测比实验省时省钱?
从时间上看,实验方法破解一个蛋白质结构,根据研究的紧急程度、样本可获得性、蛋白复杂程度等等因素,用时短则几周几月,长达数年也不稀奇。相比而言,计算预测是要快一些,毕竟AlphaFold运算训练只花费了数周,而CASP竞赛全套建模项目也只有5个月。
除了省时,也可能更省钱。
冷冻电镜、核磁共振、X射线衍射三种实验方法探测蛋白结构的原理不同,设备也全然不同。冷冻电镜是用电镜观察经特殊冷冻后的样本;核磁共振是通过分析原子对能量的吸收情况反推物质的构成;X射线衍射法是利用晶体的X射线衍射结果计算晶体中原子排布。
硬件成本上,一台冷冻电镜根据规格型号不同,设备价格约在2000万到6000万之间,这还不算运行和维护费用。
而核磁共振和晶体学的设备价格也很高昂。特别是晶体学的同步辐射设备,占地可达数平方公里,建设费用数以亿计,李赛介绍说。
陈勇是清华大学生命科学学院博士后,博士期间在中科院生物物理所研究晶体学,他表示X射线晶体学的设施建设费用可达百亿,一般由政府主导,比如中国的上海同步辐射光源。这些平台除了服务生物学,还用于材料科学、物理学等。虽然造价上天,陈勇解释,同步辐射设备通常全球共享,并不是每个研究单位都要自己有。
而如果用计算机预测蛋白结构,硬件 “主要是高性能GPU计算平台的投入”,李赛表示,一个满足学院规模计算需求的平台建设大概需要千万级别,每年维护费用相对实验设施投入较低,电费开支每年在百万。
陈勇认为,计算机预测蛋白结构的成本理论上还是有优势——虽然预测需要高性能计算机,但冷冻电镜也需要高性能计算,而且还要加上前期设备费用。
4 机器预测能否代替实验?
即使计算的成本优于实验,但有了预测还做不做实验呢?
“AlphaFold确实是有划时代意义的工作,因为过去几十年科学家们一直努力实现能不能通过计算预测。这个是计算模拟或者是预测,实验性科学永远是实验性科学,我不知道计算预测出来的结果还要实验科学去验证吗?” 陈勇说,“这是个有意思的问题,我也不知这会不会出现重复性工作,你预测出来一个,我还要去验证一下?”
对于结构生物学研究,预测并不陌生,并一直作为实验科学的辅助存在。
李赛告诉《知识分子》,在AlphaFold之前很多年就有了一些预测蛋白质结构的算法。“密歇根大学的张阳实验室在这个上面比较领先,我们有时候会用他们的服务器预测一些结构,用来和电镜结构做参照。” 至于预测与实验的结果,有时候一样,很多时候是不一样。
不过,对于从零开始的研究,预测可以帮助实验科学找找思路。陈勇表示,有许多未知的蛋白结构,在前期没有任何东西参考的情况下,“可以通过序列计算出来一个模型,然后实验性科学正好又得到了一个蛋白结构的电子密度,这样在分辨率没那么高的情况下,可能有助于我们从头建模,去搭原子模型。”
对于AlphaFold这样高准确性的算法,“我觉得这是一个蛮好的事情,非常厉害,”陈勇说,预测能辅助搭模,帮助在做表达的时候做一些优化和改造,“这样可能会加速实验性科学的脚步。”
实验受益于预测,而预测的算法是基于实验科学的结果。
或许蛋白质折叠存在某种可靠的规律,但这种规律需要参考的生理因素实在太多太多。实验科学尚在探索冰山一角,构筑在实验之上的算法能形成理论闭环吗?
“完全基于物理学和化学第一性原理的结构预测还没有出现。实验科学永远是探索未知的必要手段。”王宏伟指出。AlphaFold系统对蛋白结构精准预测的算法不是凭空成立的,而来自对17万实验确定的蛋白结构和序列的学习。
所有实验解出蛋白结构会上传PDB数据库并拥有编号。“这周放出来的晶体结构的量是两百多个,电镜(实验获得的)大概是五十多个。”陈勇介绍说。另外,数据库里约10~15%的结果来自核磁共振。
创建于1971年的全球共享的蛋白结构档案库——PDB现孜孜不倦地每周二更新。截至2020年12月2日,共收录171916个蛋白结构。
但更多的蛋白质结构仍然未知。
“对这些蛋白进行结构预测具有极大的重要性,同时预测的结构也有助于实验解析未知蛋白结构。” 李赛表示。
DeepMind表示,在未确定的蛋白质中,可能有一些具有令人兴奋的新功能,像AlphaFold这样的技术可能会帮助我们找到它们——就像望远镜帮助我们看到未知宇宙的更深处一样。
探索更多的未知结构,这大概是 AlphaFold(们)和结构生物学家的共识吧。
参考资料:
[1]High Accuracy Protein Structure Prediction Using Deep .
[2]‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures.https://
[3]RCSB PDB. #Category-analyze
[4]
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}